 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司医疗器械技术文件包括哪些?

术语“技术文件”是指医疗设备制造商必须提交的所有文档。技术文件是合格评定的要求,因此也是医疗设备批准的要求。下面我们一起来看下医疗器械技术文件包括哪些?

1. 技术文件的法规要求
a)医疗器械指令93/42 / EEC(MDD)
所述医疗器械(93/42 / EEC)的医疗器械,其中包括所谓的指定要求的基本要求。制造商在法律上有义务证明符合这些要求。通过技术文件证明了此合规性。
《医疗器械指令》(与MDR相反)没有明确说明文件本身的要求。但是,它提到:它们必须包括,例如:
- 产品的一般说明,包括计划的任何变体及其预期用途;
- 设计规范,包括将要应用的标准和风险分析的结果,以及对如果未完全应用第5条中提到的标准而满足产品所必需的基本要求而采用的解决方案的说明;
- 设计产品时将使用的用于控制和验证设计以及过程和系统措施的技术;
- 如果要将设备连接到其他设备以按预期方式运行,则必须提供证明,证明当连接到具有制造商指定特征的任何此类设备时,该设备符合基本要求;
- 根据附件一第一章第二节采用的解决方案;
- 临床前评估;
- 附件十所指的临床评估;
- 标签草案以及适当的使用说明。
- 所有这些文件都必须由制造商在合格性评估过程中提交-作为技术文件。
b)医疗器械法规(MDR)
该医疗器械法规(MDR)不只是定义了设备(附件一)的要求,同时也定义了文档本身(附件二)的要求。这必须包括(见图1):
- 设备标识(例如,使用UDI)
- 设备说明,包括变型,配置和附件
- 有可能的使用
- 标签 (包装,使用说明等)
- 有关设备设计和制造的信息
- 风险管理文件
- 验证和确认设备,从而证明设备符合一般的安全和性能要求。
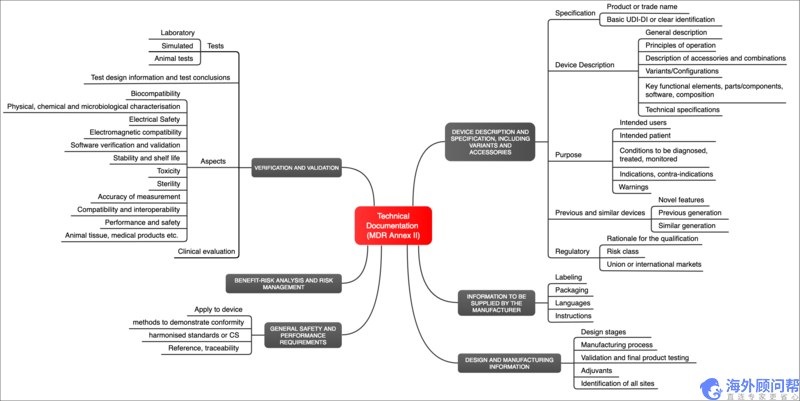
图1:MDR在附件II中指定了技术文件的要求
MDR更进一步:它包括技术文件下的售后监督及其计划和实施。它在附件三中规定了相应的要求。
(c)ISO 13485:2016年
2016年版的ISO 13485引入了医疗设备文件。该文件必须提供类似的信息:
- 设备说明
- 目的
- 标签(包装,标记,使用说明,安装和维护说明)
- 设备规格
- 其制造,包装和储存规范
- 市场监督
- 符合性证明,包括验证和确认
d)公告机构
指定机构还发布了建议,例如 NB-MED / 2.5.1。这些出版物没有法律效力。但是,在审核和审查技术文件期间仍会定期要求其内容。
e)FDA
FDA还需要详细的设备文档。它区分以下几个文件:
- 设计历史文件(DHF)
- 设备主记录(DMR)
- 设备历史记录(DHR)
2. 技术文件的内容
上述规定定义了技术文件必须包含的方面。但是它没有描述:
- 具体内容,例如软件架构或生物相容性测试必须描述什么?
- 内容在文档中的分布
- 文件结构(章节结构)
如ISO 14971,IEC 62304,IEC 60601-1和IEC 62366-1,确实给了内容详细的规格。因此,ISO 62366-1规定文档必须包括形成性评估的计划。在MDD,MDR或ISO 13485中找不到此类细化的要求。
以下思维导图提供了医疗器械制造商必须准备进行合格性评估以证明其医疗器械符合《医疗器械指令》附录I的基本要求的文档的摘要和概述。
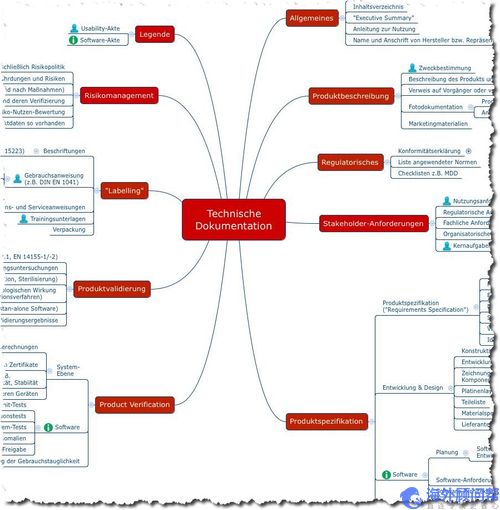
图2:具有技术文件内容的Mindmap
3. 技术文件的结构
法规要求未指定制造商应如何构造技术文件。FDA是一个例外,例如,FDA提供了章节结构,包括上市前通知的章节编号(510(k))。
a)“标准化”结构的目标
一些“协会”已经为技术文件的结构提出了建议,以试图实现以下目标:
- 制造商可以尽快了解其必须记录的内容。
- 当局和指定机构可以通过统一且合乎逻辑的结构快速找到解决办法并审查技术文件。
- 制造商在重新编写用于不同法律领域的文件上所花费的精力更少。
4. 技术文件和质量管理体系之间的相互作用
制造商必须始终草拟其医疗器械的技术文件,并将其提交给指定机构(I类器械除外)。在那里将被第一次检查。技术文件也是ISO 13485审核的主题。根据该文档,审核员不仅要评估是否符合《医疗器械指令》的基本要求,而且还要评估制造商是否按照自己的质量管理体系进行工作。最后,质量管理体系必须定义用于开发和生产医疗设备的过程(例如,开发和风险管理)。
因此,技术文件不仅是制造商必须汇编并提交给指定机构的内容,还包括在开发过程中“自动”编写的一组文档。
换句话说:如果没有一致的,符合标准的和完整的技术文件,医疗器械制造商将无法证明其医疗器械符合基本要求或其质量管理体系有效。对于大多数制造商而言,质量管理体系是进行合格评定的前提。
以上就是关于“医疗器械技术文件包括哪些?”的相关内容,了解更多请咨询海外顾问帮。
【海外顾问帮】是协助国内企业和个人跨境发展一站式的服务中心,协同全球专家顾问,坚持透明服务,打破跨境壁垒,为海外产品认证提供一站式服务,咨询电话: 400-106-2206。

医疗产品CE认证
直连专家








