 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司欧盟MDR常见问题解答

欧盟MDR是什么意思?
EU MDR是由欧洲议会和欧盟理事会于2017年发布的《欧盟医疗器械法规2017/745》。欧盟MDR法规的目的是确保在欧盟成员国生产或供应给欧盟成员国的医疗器械的高标准安全性和质量。

该监管框架旨在更好地识别医疗设备,并通过欧盟数据库(Eudamed)标准化数据和技术进步。欧盟MDR法规旨在成为医疗器械的法规框架,既可以持续确保健康与安全,又可以鼓励创新。
什么是MDD和MDR?
欧盟医疗器械指令(MDD)已经实施了将近25年,之后于2017年发布的新的《欧盟医疗器械法规》(MDR)取代。该先前的指令构成了欧盟医疗器械框架,但是不包括体外诊断医疗设备。
但是,已确定必须对这些指令进行根本修订,以建立一个更强大,透明,可持续和可预测的监管框架。目的是提高医疗设备的整体健康和安全水平,同时仍支持行业创新。
欧盟MDR何时发布?
2017年4月4日,有关欧洲医疗器械的理事会负责制定新法规,而欧盟MDR法规于2017年5月5日发布。公司要遵守新法规,过渡期为三年MDR欧盟法规。
欧盟MDR何时生效?
尽管公司可以立即开始遵守新法规,但从欧盟MDD指令到欧盟MDR法规的过渡期为四年,到2021年5月26日结束。在此日期之后,任何新的医疗器械都需要获得新的欧盟MDR法规认证。现有的MDD认证医疗设备还有一个过渡期,直到2024年5月,才能更改其技术文档以符合新的MDR EU法规。
所有现有的MDD认证设备必须在哪个日期获得MDR认证?
根据新的MDR认证的所有MDD认证设备的最终批准日期为2024年5月25日。如果在此日期之前医疗器械符合EU MDR法规,则可以通过MDR认证,但并非如此。如果其MDD证书仍然有效,则为必选。如果MDD证书在2024年5月25日之前到期,则需要根据MDR重新认证此类医疗设备。
根据欧盟MDR进行最终认证的最后日期是2024年5月25日–此日期之后,所有投放市场的设备都必须获得欧盟医疗设备法规的认证。

什么是欧盟MDR合规性?
欧盟医疗器械法规已经建立了一个独特的器械识别(UDI)系统,该系统类似于美国食品药品管理局(FDA)的系统。如果公司打算在欧盟市场上提供或分发医疗设备,则这些产品上的标签将需要符合此新的UDI系统。对于打算在欧洲销售产品的医疗设备公司,必须遵守新的MDR法规。
什么是MDR认证?
医疗器械的MDR认证可证明该器械符合欧盟医疗器械的所有法规要求;认证带有CE标志。
为了使医疗器械获得认证,您的公司必须实施质量管理体系(QMS)。许多公司使用ISO 13485作为实施此QMS的方法,因为这是欧盟协调名单上唯一的QMS标准,因此,由于它与MDR EU法规有关,因此是实施QMS的最佳方法。
与MDD相比,欧盟MDR有哪些变化?
欧盟医疗器械法规(EU MDR)取代了之前的欧盟医疗器械指令(EU MDD)。特别是,新的欧盟MDR(2017/745)修改了之前的指令2001/83 / EC,并废除了理事会的指令90/385 / EEC和93/42 / EEC。
以下是旧的欧盟MDD与新的欧盟MDR之间的六个主要区别:
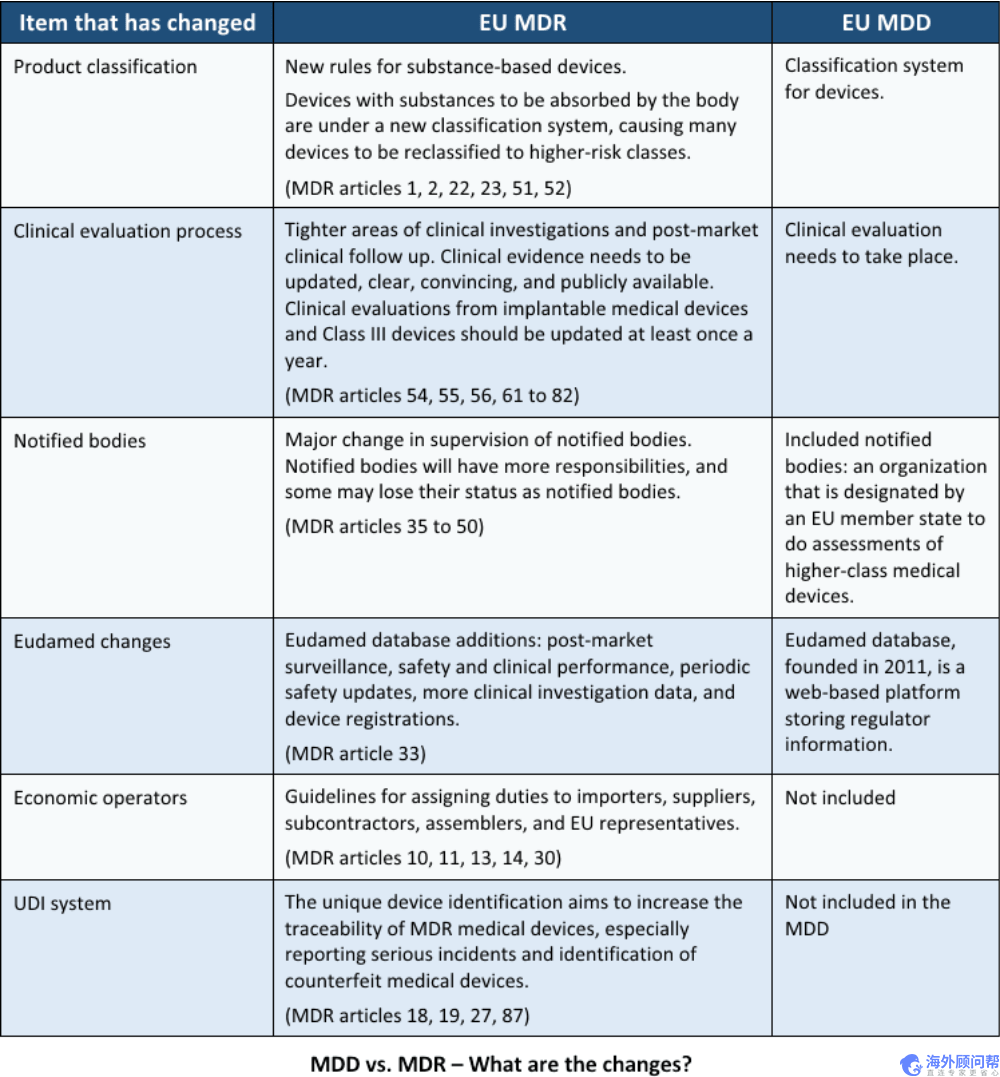
什么是MDR和IVDR?
欧盟MDR法规未纳入体外诊断法规(IVDR);此IVDR设备受单独的欧盟法规2017/746管辖。但是,MDR法规的第1条(第7点)阐明了IVDR法规与欧盟MDR法规之间的关系:如果设备同时具有IVDR组件和其他医疗设备组件,则任何设备的IVDR部件均受2017年管辖/ 746法规,但设备的其余部分受MDR EU法规覆盖。
体外医疗设备是旨在用于诊断,监视或测量人体外部样本的兼容性的设备。新的欧盟MDR法规包括对此类产品的要求。
MDR技术图
与EU MDD一样,所有带有CE标记的医疗设备产品都需要创建技术文件,以证明该设备符合EU对CE标记产品的所有要求。新的欧盟MDR附件II包括对内容,结构或技术文件中需要包含的内容的修订要求。
根据欧盟MDR附件II,技术文档必须清晰,有条理,易于检索且明确。技术文件中要包括的元素是:
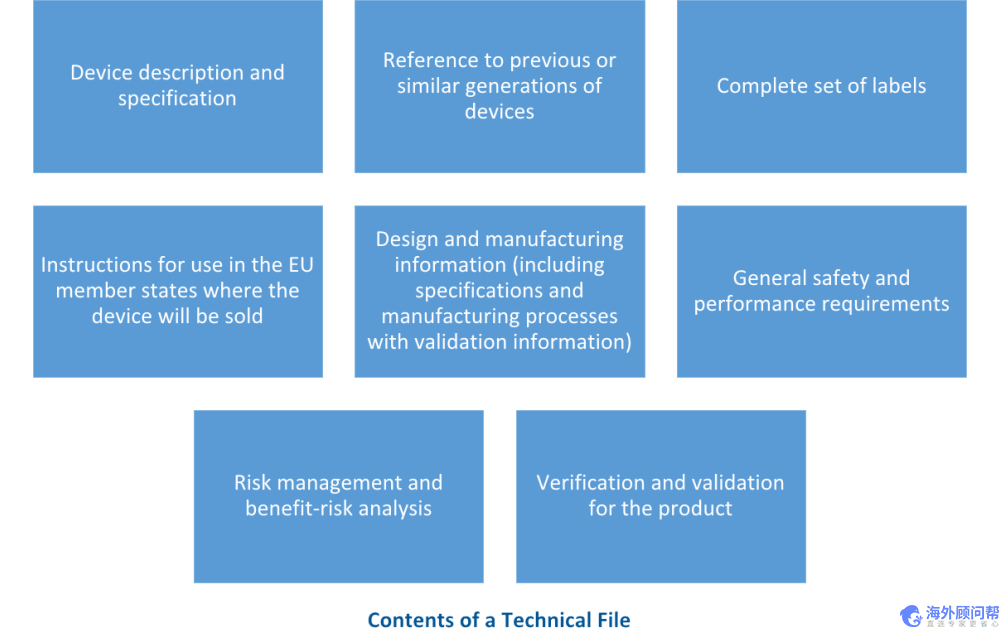
此做法与ISO 13485:2016第4.2.3条相符,该条要求设备制造商创建技术文件或医疗设备文件。
什么是第一类医疗设备?
I类医疗设备是被认为是非侵入性的医疗设备,包括以下类别:无菌,测量或可重复使用的手术设备。某些I类设备可以进行自我认证,而无需通知机构参与;但是,您仍然必须实施QMS。
医疗设备有哪些例子?
欧盟MDR法规在附录VIII中包括五类医疗设备。本附件根据22条规则对产品进行分类。以下是一个简短的摘要,每个示例都有一些示例:
I级(低风险):非无菌设备或没有测量功能的设备。这些设备可以自我认证。例如:可重复使用的手术设备(例如手术刀),听诊器,轮椅。
I类特殊功能(低/中度风险):无菌设备或具有测量功能的设备。不符合其他类别要求的非侵入性设备,以及打算暂时使用的侵入性设备。例如:再加工设备,测量设备,无菌交付的设备。
IIa类(中等风险):打算短期使用的侵入性设备。例如:牙齿植入物,气管切开管,注射器,X射线设备。
IIb级(中/高风险):旨在长期使用的侵入性设备。例如:植入式节育器,血袋。
III类(高风险):打算长期使用的可植入医疗设备。例如:起搏器,乳房植入物,椎间盘植入物,药物涂层支架。
什么是医疗保健中的MDR?
欧盟MDR法规旨在提高欧洲医疗器械的安全性和性能,因此,旨在为患者和这些医疗器械使用者的健康提供高水平的保护。为此,欧盟MDR法规为医疗设备的质量和安全设定了高标准,以更好地解决医疗设备的安全问题。
那么,欧盟MDR与医疗保健有什么关系?欧盟医疗器械法规正在建立一个框架,以确保制造或进口到欧盟的医疗器械达到这一高水平的安全性和质量。通过提高医疗设备的安全性和性能,欧盟MDR法规旨在提高医疗保健的安全性。
【海外顾问帮】是协助国内企业和个人跨境发展一站式的服务中心,协同全球专家顾问,坚持透明服务,打破跨境壁垒,为医疗产品CE认证提供一站式服务,咨询电话: 400-106-2206。








