邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司

“应加强包括监管指定机构,合格评定程序,临床调查和临床评估,警惕性和市场监督在内的现行监管方法,以进一步加强。此外,应引入更多条款以确保医疗器械的透明度和可追溯性,以改善健康和安全性。” – 2017/745号法规
| 医疗器械指令 | 医疗器械法规 |
| 93/42 / EEC(MDD)-43页 90/385 / EEC(AIMD)-20页 |
法规EU 2017 / 745-177页 废除理事会指令90/385 / EEC和93/42 / EEC(MDD + AIMD) |
| 23条,十七附件 | 123条,十七附件 |
| 分类规则总数:18 | 分类规则总数:22 |
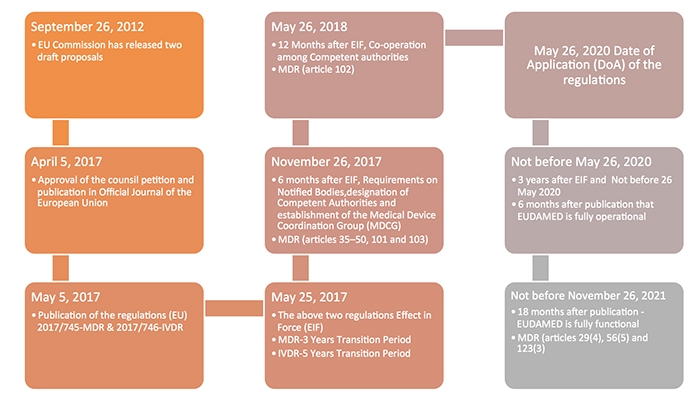
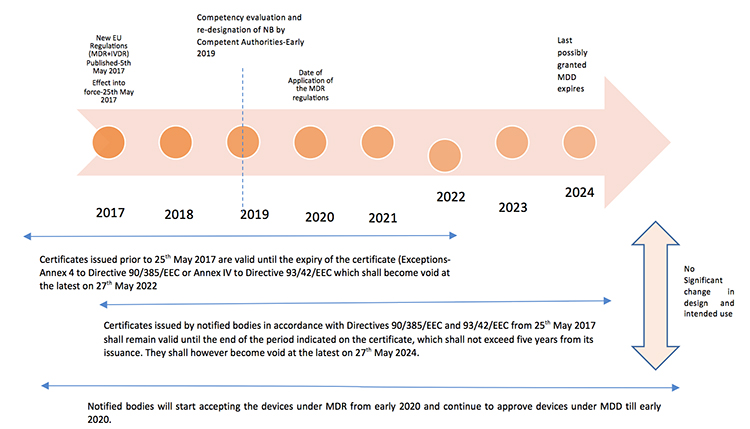
过渡时间表和证书的有效性
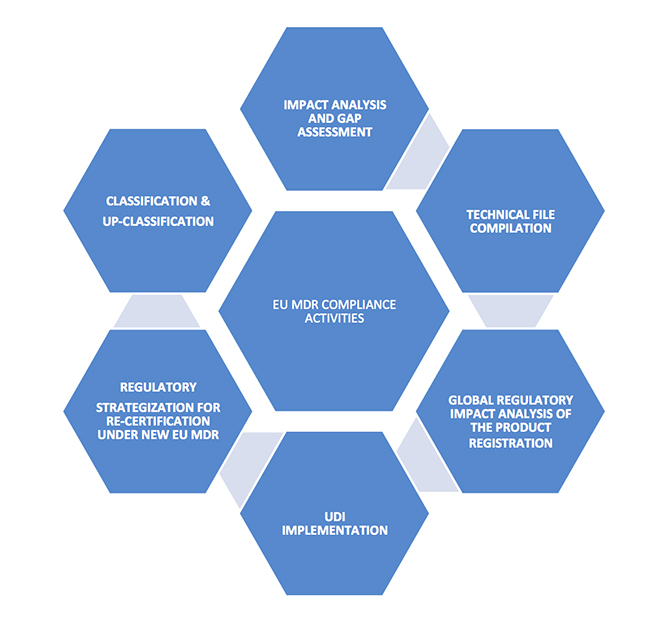
了解MDR
1. 考虑设备的分类;完成您的产品组合
如果您的设备当前是医疗设备的附件,具有美学目的或其他非医疗目的的产品,包含药物的临界产品,组合产品或软件产品,则应评估新的医疗设备法规用于设备的分类和上分类。
设备的分类和上分类对制造商有重大影响,因为欧盟MDR规定了更严格的合规性,安全性和功效要求。由于合规成本高昂,这将在最终确定当前和将来的产品组合中扮演关键角色。任何更改都应从管理层开始,包括决定欧洲市场产品组合,评估基于CE认证的全球产品批准和市场准入的影响的关键决策者。
在规定的时间表内不遵守新要求将导致失去在欧洲销售产品的许可。
2. 确定主要的合规要求
熟悉新的MDR术语:医疗设备协调小组(MDCG),一般安全和性能要求,通用规范,唯一设备标识,虚拟制造商。
对于根据新规则新分类为医疗器械的产品,将加强对指定机构的审查。根据重新分类和现有产品的新法规,应准备好新文档以进行CE认证。应制定的一些关键技术文件包括:
- 风险分析文件
- 临床数据可用性分析和计划
- 一般安全和性能要求
- 通用规格推导
- 所有相关的质量检查SOP
- 根据权利要求的设备性能评估测试
- 标签更新
- 符合ISO 13485产品制造和认证要求
- PMCF计划