 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司2017年的《医疗器械条例》(MDR)增加了对临床数据记录以支持许可证申请的要求

在1993年之前,医疗器械的临床安全性和性能文档受到的关注远少于其制造和质量文档。自1993年6月以来,为提高医疗设备的安全性进行了一系列监管工作。
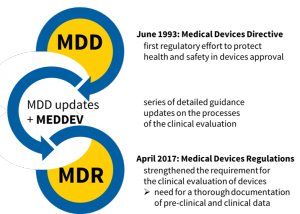
MDD =医疗器械指令(93/42 / EEC);MDR =医疗设备法规(2017/745)
2017年4月,MDR发布,并针对所有类别的医疗器械进入欧盟市场进行了法定的医疗器械临床评估。尽管期望从MDR获得新的指南,包括有关特定类型设备的指南,但与临床评估有关的MEDDEV指南在很大程度上仍然有效。MDR加强了对医疗器械进行临床评估的要求,尤其是需要对与器械的安全性和性能有关的临床前和临床数据进行全面记录的要求。记录所需的临床前和临床数据的数量在很大程度上取决于与使用该设备有关的健康和安全风险。引入了几种新的文档类型:
- 临床评估报告(CER)
- 临床发展计划(CDP)
- 临床评估计划(CEP)
- 上市后临床随访(PMSF)计划
- PMSF报告
- 失效模式和影响分析(FMEA)
- 临床调查计划
- 临床调查报告
并非所有设备都需要所有这些,但是现在欧洲所有医疗设备许可证申请都必须使用CER。对于监管机构(公告机构),CER是评估医疗器械在临床上的适用性的关键要素。
关于医疗器械临床评估要求的详细信息首先在MDD中提供,但有关CER内容的详细信息首先在EU MEDDEV准则2.7 / 1 rev 4(2016)中出现。它记录了支持许可申请的临床证据,并且在批准后会定期进行更新,以使该设备能够继续投放市场。
CER描述了医疗器械的临床评估,根据《 2017年MDR》,“……应是彻底和客观的,并应考虑有利和不利的数据。其深度和程度应与所涉设备的性质,分类,预期目的和风险以及制造商有关该设备的主张相称并适当。”
CER是独立文件,并作为技术文件的附件提交。它包含了:
- 使用制造商在上市后的临床随访过程中积极获取的证据,评估设备安全风险和预期的临床性能的证据,并在更新中提供有关其在现场安全性和性能的其他临床数据;
- 定期更新有关出版文献的评论;
- 根据设备的类别和类型,可以从设备的临床调查获得数据。
欧盟MEDDEV准则2.7 / 1 rev 4(2016)仍然是当前公认的医疗器械临床评估流程指南,包括有关CER的结构和内容的指南。它提出了CER的目录,该目录现已被广泛接受为标准。
欧盟法规536/2014的后果
这项新的《欧洲临床试验法规》重新定义并加强了涵盖欧盟(EU)内任何地方进行的临床试验的基本法规框架。该要求最初计划于2018年生效,但是数据库和上传门户的创建被延迟了,因此很可能在2020年之前不会实现。
新法规的目的是创造一个有利于在欧盟内进行临床试验的环境,并为参与者提供最高的安全标准。它还要求在整个欧盟范围内保持一致,并制定新的临床试验行为规则,并通过要求透明授权,进行试验和结果来转变每个临床试验的公开信息水平。
新法规对临床文档义务产生重大影响的具体规定如下:
- 为了增加透明度,如果临床试验数据已经记录在可公开访问且免费的数据库中,则该临床试验数据将来只能在支持该临床试验申请的情况下提交,该数据库是该数据库的主要或合作伙伴注册表或数据提供者,世界卫生组织(WHO ICTRP)的国际临床试验注册平台。相关数据有望通过EMA的欧洲门户网站提交到欧洲数据库。
- 申办者将被要求将临床试验结果的摘要提交给可公开获得的欧洲数据库,并提供外行可以理解的随附摘要。只有在出于科学原因无法在规定的时间表内提交结果摘要的情况下,才允许延迟,例如,当临床试验仍在第三国进行且该部分试验的数据不可用时–统计分析不相关。在这种情况下,申办者必须证明延缓试验方案的合理性,并指定何时提交结果。
- 公众将能够访问所有临床试验的详细信息,包括试验的主要特征,招募的开始和结束,试验的结束日期以及对试验的实质性修改。这些细节将从发生审判时开始,并在发生时予以公布。试验结束后12个月将发布结果摘要和常规摘要。对于欧盟市场许可申请中包含的试验,临床研究报告还将在授予市场许可的程序完成或撤回申请后30天发布。

医疗产品CE认证
直连专家








