 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司官方解读:医疗器械法规MDR

自2017年5月25日起,《医疗器械法规MDR》和《体外医疗器械法规IVDR》 生效。这些法规替代了欧盟指令(MDD,IVDD和AIMD)。自2020年5月26日起,MDR应于2022年5月26日适用IVDR。

医疗器械指令:什么保持不变?
这些变化是重大的。但是,一些核心概念保持不变:
- 没有监管机构/机构:将不会有欧洲机构授予与药品类似的欧洲药品管理局EMA或药品,医疗器械等获得FDA许可的市场准入。
- 遵循合格评定程序后,制造商仍需声明合格(带有CE标志)。合格评定程序仍取决于设备的类别。除I类设备外,指定 机构仍必须参与。
- 该分类基本上是不变的。异常在下面提到。
- 统一的标准仍然是证明合规性的一种方法。但是,欧盟还引入了“通用规范”的概念(见下文)。
- 非欧盟制造商可以进口医疗设备。前提条件之一仍然是欧盟代表。
MDR:有什么新东西?
《医疗器械法规》是一份涵盖550多页的文档,是《医疗器械指令》 MDD的倍数。MDR引入了许多新要求,并具体化了MDD的要求。
- 基本要求
的基本要求-现更名为“基本安全和性能要求” -是更具体的。因此,技术文档受到更严格的监管。MDD和MDR要求之间存在比较。
- UDI和标签
与FDA一样,制造商现在必须分配唯一的设备标识(UDI)。该UDI由设备标识符和生产标识符组成。UDI并不是对标签要求的唯一更改。
- 新角色
制造商必须雇用(小公司除外)“负责法规遵从的人员”。其他“经济运营商”包括分销商,欧盟代表和进口商等角色。
- EUDAMED
不仅必须将UDI数据提交到EU数据库(EUDAMED),而且还必须(取决于类别)提交上市后数据(例如,“定期安全更新报告”)。
- 临床评估和PMCF(上市后临床随访)
MDD仅在一页上讨论临床评估,而上市后临床随访(PMCF)则根本没有。MDR在几篇文章和详尽的附件中准确描述了各自的要求。
- 上市
后监视现在对MDR中几乎未解决的上市后监视进行了详细监管。
- 投放市场的合格评定程序
合格评定程序已更改:不再有与MDD附件VI相当的程序。另外,对于高风险产品(某些IIa类和大多数III类产品),必须遵循附加程序,指定机构必须咨询专家组/专家组(审查)。
- 分类
现在,分类规则考虑到了有源可植入设备,纳米材料和引入人体的物质。新的分类规则11专门针对软件-预计将进行重大重新分类。
- 通用规范
如果缺乏统一标准,欧盟委员会有权采用“通用规范”(CS)。到目前为止,还没有统一标准,也没有发布任何CS。
欧盟法规与欧盟指令
同样重要的是要理解,欧盟法规自发布之日起对所有欧盟国家有效。相反,欧盟指令必须首先在国内法中执行。
仍然会有国家法律,例如定义罚款。但是,MDR规定了制造商和进口商必须遵守的基本要求。
瑞士是非欧盟国家,仍是欧盟体系的一部分。
与美国FDA法规的主要区别
新医疗器械法规的要求与FDA要求非常接近。但是,已经获得FDA许可的制造商必须考虑:
- 成功通过合格评定程序是前提条件。
- 除了I类产品,这还意味着要有认证机构和符合ISO 13485的质量管理体系的参与。
- 制造商通常必须通过应用诸如IEC 62304,IEC 62366和ISO 14971的协调标准来证明其符合性。幸运的是,这些标准与指导文件中所述的FDA要求非常相似。
- 显然,医疗设备报告是不同的。这不仅适用于表格和收件人。
- 欧盟对“经济运营商”(如进口商,分销商,欧盟代表)和“负责法规要求的人员”有专门的要求。
- 关于临床评估和上市后临床随访的要求不同(部分更高)。
医疗器械法规(MDR)概览
《医疗器械法规》 MDR由10章组成,总共123条。MDD包含23条文章。
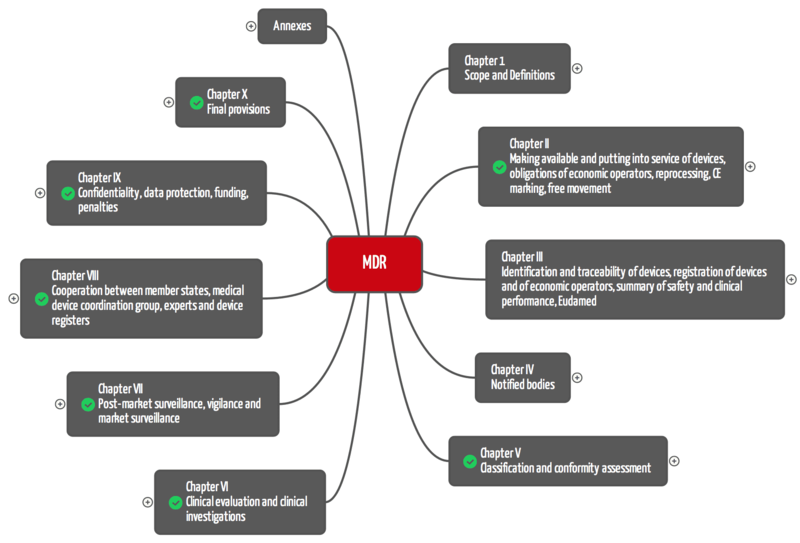
对于制造商,最相关的章节为:
- 第二章:设备的可用和投入使用,经济运营商的义务,后处理,CE标志,自由移动
- 第五章分类与合格评定
- 第六章:临床评价与临床研究
- 第七章上市后监督,警惕和市场监督
此外,MDR具有22个附件:
- 一般安全和性能要求
- 技术文件
- 上市后监督的技术文件
- 欧盟符合性声明
- CE合格标志
- 注册时要提交的信息……
- 指定机构应满足的要求
- 分类规则
- 基于质量管理体系的合格评定和技术文件的评定
- 基于型式检验的合格评定
- 基于产品合格性验证的合格性评估
- 认证机构签发的证书
- 定制设备的程序
- 临床评估和上市后临床随访
- 临床研究
- 第1条第(2)款中没有预期医疗目的的产品组列表
- 相关表
MDR:时间轴
MDR于2017年2月出版,自2017年5月25日起,MDR“生效”,三年后适用。
以上就是关于“医疗器械法规MDR”的相关内容,了解更多请咨询海外顾问帮。
【海外顾问帮】是协助国内企业和个人跨境发展一站式的服务中心,协同全球专家顾问,坚持透明服务,打破跨境壁垒,为医疗产品CE认证提供一站式服务,咨询电话: 400-106-2206。








