 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司FDA 510(k)怎么做?在哪办理?代理机构有哪些?

最近经常有人过来咨询FDA 510(k),为了帮大家解决疑惑,今天小编就给大家整理了一下问的最多的一些问题,希望对你们有帮助。如果看了下面的内容还是有不懂的地方,或者是想办理FDA认证,可以咨询我们的在线客服,会有专家顾问帮您解答。

FDA 510(k)
根据风险等级的不同,美国食品药品监督管理局(FDA)将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),Ⅲ类风险等级最高。
如果您计划进入美国市场,除了质量管理体系需要符合美国的 GMP 要求,即通常所说的 QSR820,还需要进行企业注册(Registration)和产品列名(Listing)。其中,绝大部分 I 类器械可直接进行产品列名,极少数产品还豁免 GMP;大部分的 II 类器械,以及少量的 I 类 和 III 类器械,需要向 FDA 提交 510(k)申请,获取批准函(Clearance)后,再进行产品列名;剩下的 III 类器械则需要向 FDA 递交 PMA(Premarket Application)申请,获得批准函后再进行产品列名上市。
1. 美国 FDA 认证是什么?
美国食品和药物管理局(Food and Drug Administration)简称 FDA,FDA 是美国政府在健康与人类服务部 (DHHS) 和公共卫生部 (PHS) 中设立的执行机构之一。作为一家科学管理机构,FDA 的职责是确保美国本国生产或进口的食品、化妆品、药物、生物制剂、医疗设备和放射产品的安全。换言之,食品,化妆品,药物,生物制剂,医疗设备和放射产品在美国进行销售必须要通过 FDA 注册批准。
医疗器械 FDA 认证是医疗器械行业对医疗器械进入美国市场之上市前注册流程的习惯性叫法。所有的医疗器械,企业都需进行企业注册(Registration)和产品列名(Listing)。
企业在计划进入美国市场前,需仔细评估针对自己产品相关的法规和具体要求(包括不同的美国产品标准要求)。
2. 什么是 510(k)?
FDA 510(k): Premarket Notification 上市前通告 PMN,510(k)是向美国 FDA 提交的上市前文件,用以证明所销售的设备与合法销售的产品实质性等同(Substantially Equivalent),即基本上等同于合法销售的设备。部分的 II 类器械,以及少量的 I 类和 III 类器械在上市前都需要向 FDA 提交510(k)申请。
3. 哪些情况下需要提交 FDA510(k)?
- · 首次将一种医疗器械引入美国市场
- · 改变已经入市的器械的使用目的
- · 对已经入市的器械进行改变或更新(这种变更或更新会影响器械的安全或有效性,这种改变或更新包括设计、材料、化学成分、驱动力、生产流程或者预期用途)
4. 谁必须要申请 FDA510(k)
FD&C Act 的第 510(k)规章中并没有特别指出谁必须申请 510(k)——任何人都可以申请。但是,他们指定了哪种行为,例如把器械引入美国市场,要求 510(k)申请。基于指定的行为,必须向 FDA 递交510(k)的有:
(1) 把器械引入美国市场的生产商;
(2) 把器械引入美国市场的研发设计者;
(3) 改变器械或器械标签的再包装者;
(4) 把器械引入美国市场的外国厂家/出口商或外国厂家/出口商的美国代理方
5. FDA 510(k)怎么做?
510(k)申请流程图
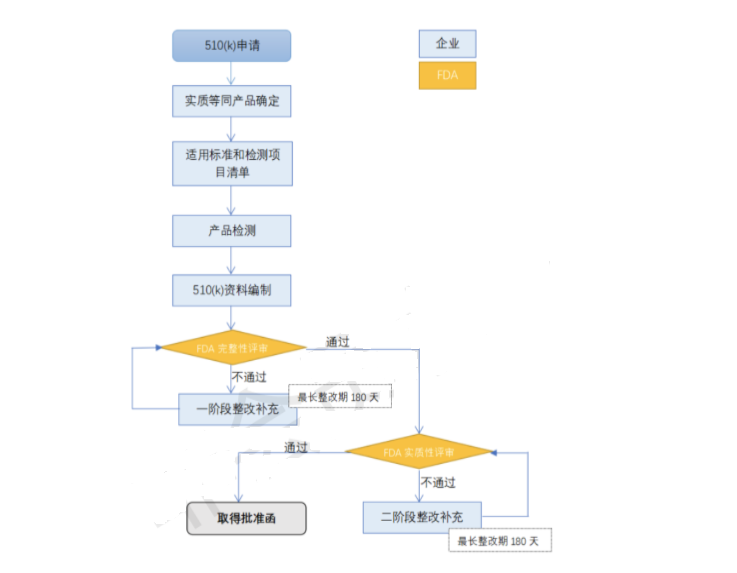
6. FDA 510(k)在哪办理?
海外顾问帮作为全球化的医疗器械法规专业技术服务商,可提供服务:
(1)确定您的医疗器械在美国的具体分类及风险级别;
(2)辅导企业准备 510(k)申请所需的资料;
(3)确定检测标准及适用项目,推荐测试机构,对检测报告进行审查;
(4)编撰 510(k)申报文件;
(5)辅导企业进行小企业资质申请;
(6)跟踪 510(k)文件评审进度;
(7)辅导企业整改发补问题;
(8)进行企业注册和产品列名。
以上就是关于“FDA 510(k)怎么做?在哪办理?代理机构有哪些?”的相关内容,了解更多点击咨询。

美国FDA认证
直连专家








