 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司医疗器械MDR产品分类变化有哪些?

现在打算将自己的产品放入欧盟国家的制造商,必须要遵守欧盟国家的法律法规,否则你的产品无法在欧盟国家市场进行销售,在入欧盟市场之前首先你得将你的医疗器械MDR产品分类,那么目前新法规里面医疗器械MDR产品分类变化有哪些?小编带你看看吧!

一、医疗器械MDR的介绍
现在欧盟官方期刊正式发布了欧盟医疗器械法规《MDR》。 MDR将代替EEC医疗器械指令。根据MDR的要求,MDR将于2017年5月26日正式生效,并于2020年5月26日正式取代MDD、EEC 和aimdd 。引入MDR后,在3年的过渡期间也可以按照MDD和AIMDD申请CE证书,以维持证书的有效性。 根据规定,过渡期间NB颁发的CE证书将继续有效,但自其交付日起有效期不超过5年,于2024年5月27日失效。
二、医疗器械MDR产品的分类变化
从MDD到MDR,仪器仍分为4类,器械仍分为四大类:I类、IIa类、IIb类、III类。
MDD中与分类相关的是93/42/EEC的Annex IX和相应的指导方针MEDDEV 2. 4/1 Rev. 9; 在新的MDR中,Article51和Annex VIII详细介绍了产品分类。 主要变化从MDD的“18条”变成了MDR的“22条”。
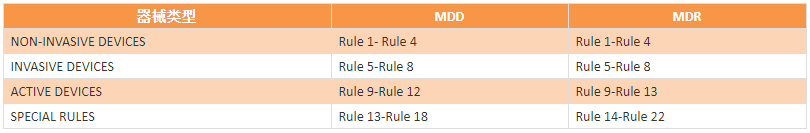
1- 4、非无源设备非侵入式设备。
3、在体外直接从人体或人类胚胎添加用于体外使用的人体细胞、组织、气管,移植或注入体内,这种仪器属于III级。
5-8、非活动设备侵入性设备。
在原有的基础上,增加了主动植入物及其相关附件、乳房植入物或**网状织物的修补、关节置换物的完整或部分、与脊柱直接接触的椎间盘置换植入物为类。
9-13、活动设备活动设备。
添加了“针对治疗目的释放电离放射线的激活设备”和“控制、监测或直接影响激活嵌入设备的设备”,这两种设备都是II b型。
新添加的用于提供诊断或治疗目的的确定和生理过程的监测的软件,均为a级; 其他软件类为I类。

欧盟授权代表
14 -22、特殊规则特殊规则
进一步完善了“由人体血液或血浆衍生的医疗产品”分类的要求。
18、进一步完善“利用经惰性或惰性处理的来自人体或动物的组织或细胞或其他衍生物制作的仪器”的分类要求。
19、添加对纳米材料设备的分类要求。
20、追加了根据吸入方式的身体端口相关的侵入器具的分类。
21 、添加了将人体吸收性物质导入人体的器具。
22、增加了具有综合诊断或合并诊断功能的能动治疗器具的分类。
此外还删除了MDD中对血袋的单独分类。
三、MDR的主要变化
1、扩大了应用范围;
2、提出了新概念和仪器的定义;
3、对医疗器械的分类进行了细分;
4、完善了仪器的通用安全和性能要求;
5、加强对技术文件的要求;
6、加强设备上市后的监管;
7、完善临床评价的相关要求;
8、建议建立和使用Eudamed数据库;
9、提出设备可追溯性(UDI);
10、对nb提出严格的要求。
四、MDR适用范围扩大
1、新的医疗器械法规MDR包括一般医疗器械MDD指令所涵盖的医疗器械和AIMDD所涵盖的许多产品;
2、具有控制或支持用于设备清洁、消毒或灭菌的设备功能的设备也适用于MDR法规;
3、Annex XVI列举的意外医疗目的产品也适用于MDR法规,如瞳美、面部填充或注射、纹身、皮肤改善和美容等产品。
五、仪器的通用安全和性能要求
从MDD的annex 1基本要求进一步完成,从原来的13个条款增加到现在的23个条款, 同时,MDD中的Article 在MDR中作为另一章要求多个性能要求。
六、MDR技术文件的基本内容的设备说明和性能指标
技术文件的要求在MDR中添加了对技术文档内容的要求; 另外,明确指出上市后的监管计划和安全性更新报告(PSUR)都是技术文件的一部分,要求根据上市后监管系统收集到的资料适当更新技术文件中。
医疗器械MDR产品分类变化有哪些?以上就是小编整理的资料,打算将自己的产品放入欧盟国家的制造商,赶紧拿着小本儿记下来吧!如果您还有不懂的地方,可以点击咨询。

医疗产品CE认证








