 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司医疗器械FDA分类标准

希望在美国营销和销售医疗器械的公司必须获得美国食品和药物管理局 (FDA) 的批准。获得批准的第一步是对您的医疗器械进行分类。这是关键的一步,因为它决定了 FDA 批准所需的上市前提交和申请的类型。它还可以帮助您在整个产品生命周期中进一步定义法规要求,并设定切合实际的商业化里程碑。今天我们就来看下医疗器械FDA分类标准。

FDA分类的医疗设备集成,作为I类,II类或III类。这种三层分类是基于产品的预期用途、使用适应症和它带来的风险。
预期用途描述了医疗器械的一般用途或功能
- 使用适应症描述了医疗器械将诊断、治疗、预防或治愈的疾病或病症。他们还描述了目标患者群体。
- 不同的监管控制(即,一般、特殊或上市前批准)分配给每个分类,以合理保证设备的安全性和有效性。随着设备从 I 类发展到 III 类,监管控制的数量会增加。
一般控制适用于 I、II 和 III 类医疗器械,除非法规豁免。这些控制包括:
- 设备制造商注册(例如,510(k) 上市前通知和设备列表)
- 掺假设备(不适合使用)
- 贴错标签的设备(提供虚假或误导性标签)
- 被禁止的设备
- 通知和其他补救措施(即通知、更换、退款、补偿或强制召回)
- 器械的记录和报告(即不良事件、器械跟踪、唯一器械识别系统或移除和更正报告)
- 关于控制供人类使用的器械(即定制器械、受限器械、良好生产规范 (GMP)、研究用器械、被视为新药的器械或人道主义器械)的一般规定
- 特殊控制适用于 II 类设备。它们通常是特定于设备的,包括:
- 性能标准(例如,设计特性或规格)
- 上市前数据要求
- 上市后监督
- 患者登记
- 特殊标签要求
- 指导文件
- III 类设备需要上市前批准 (PMA)。作为 PMA 申请的一部分,实验室和临床试验数据形式的大量科学证据必须提交给 FDA,以证明该设备的安全性和有效性。
如果您计划向美国市场推出不需要 PMA 的新设备,您很可能需要提交510(k) 上市前通知以获得 FDA 批准。510(k) 是一种更快、更便宜的提交途径,如果您的设备实质上等同于已获得 FDA 批准的设备,则可以使用该途径。510(k) 提交应包括有关您的医疗设备的详细技术、安全和性能信息。
| 设备分类 |
风险 |
监管控制 |
FDA 提交/申请类型 |
| 一级 |
低到中等 |
一般的 |
510(k) 上市前通知 *此类中的大多数设备免于 510(k) |
| 二级 |
中到高 |
一般和特殊 |
510(k) 上市前通知 |
| 第三类 |
高的 |
一般和上市前批准 (PMA) |
上市前批准 (PMA) |
* I、II 和 III 类设备的要求在Title 21 CFR Part 820和ISO 13485中进一步定义。
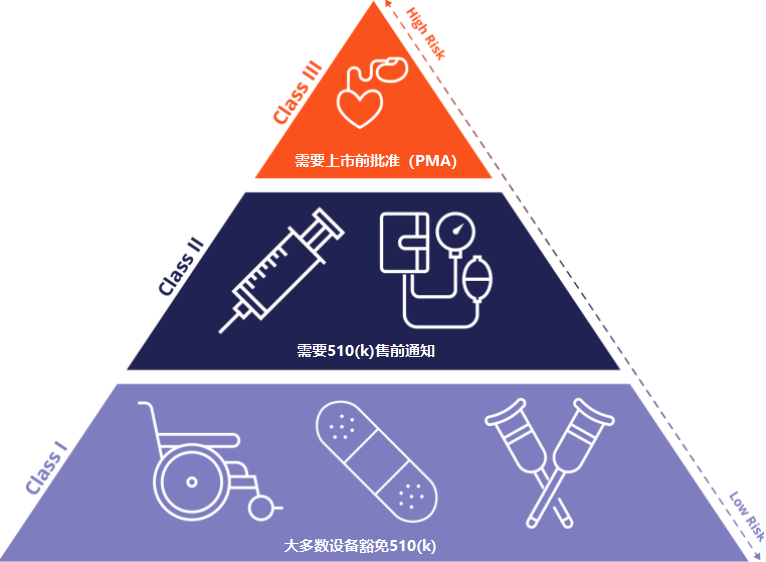
FDA医疗器械类别之间的差异
I 类设备
I 类设备对患者和/或预期用户构成低到中等风险。如今,几乎 50% 的医疗器械都属于 I 类。此类别中的设备受一般监管控制;然而,大多数免于 510(k) 上市前通知,并且在美国销售这些设备之前不需要 FDA 批准。I 类设备的示例包括绷带、一次性手套、压舌板、医用温度计和听诊器。
II 类设备
II 类设备对患者和/或预期用户构成中到高风险。今天,超过 40% 的医疗器械属于 II 类。此类别中的设备受一般和特殊监管控制的约束,大多数需要 510(k) 上市前通知才能获得 FDA 批准。II 类设备的示例包括导管、注射器、隐形眼镜和妊娠试验套件。
III 类设备
III 类设备对患者和/或预期用户构成高风险。只有 10% 的医疗器械属于这一类别。III 类设备需要申请 PMA,这需要大量临床数据来支持其安全性和有效性。这些设备通常有助于维持或支持生命,也可以植入。III 类设备的示例包括除颤器、起搏器、乳房植入物和植入假肢。
以上就是关于“医疗器械FDA分类标准”的相关内容,如果您想了解更多信息或办理医疗器械FDA认证请咨询在线客服或致电:400-106-2206。

美国FDA认证








