 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司MDR标签是什么意思?

MDR标签是什么意思?
根据欧盟医疗器械法规 (MDR)(第 2 条)中的定义,“标签”是指出现在器械本身、每个单元的包装上或产品包装上的任何书面、印刷或图形信息。多个设备。贴标签过程的目的是识别医疗器械及其制造商,并传达有关安全、使用和性能的基本信息。它适用于医疗设备的用户,包括专业人士和消费者,以及相关第三方。

众所周知,MDR 给医疗器械制造商带来了许多挑战。其中之一是标签,新要求要求在医疗器械标签上注明各种信息。
一、新MDR下的标签要求
与 MDD 93/42/EEC 相比,欧盟 MDR 下的标签需要更多信息,因为设备安全性和临床有效性数据需要与用户(医务人员和患者/最终用户)透明共享)。欧盟 MDR附件 I 第 III 章“一般安全和性能要求”中涵盖了有关随医疗器械提供的信息的所有要求。
尝试遵守欧盟 MDR 标签要求时可能会遇到两个问题。一是确保涵盖所有必要的符号和信息。另一个是标签的大小。由于需要更多的符号和数据,最大的挑战将是如何将它们全部放在标签上。在标签设计过程中,请记住以下几点:标签和说明的媒介、格式、内容、易读性和位置必须与预期用户的技术知识、经验、教育或培训相匹配。此外,使用说明必须以预期用户易于理解的术语编写,并在适当的情况下补充附图和图表。
很高兴知道您可以选择格式;标签可以以人类可读的格式提供,并且可以用机器可读的信息来补充。
二、标签上的新元素
每个医疗器械都必须有说明它是医疗器械。如果设备仅用于临床研究,则标签必须包含“专用于临床研究”字样。
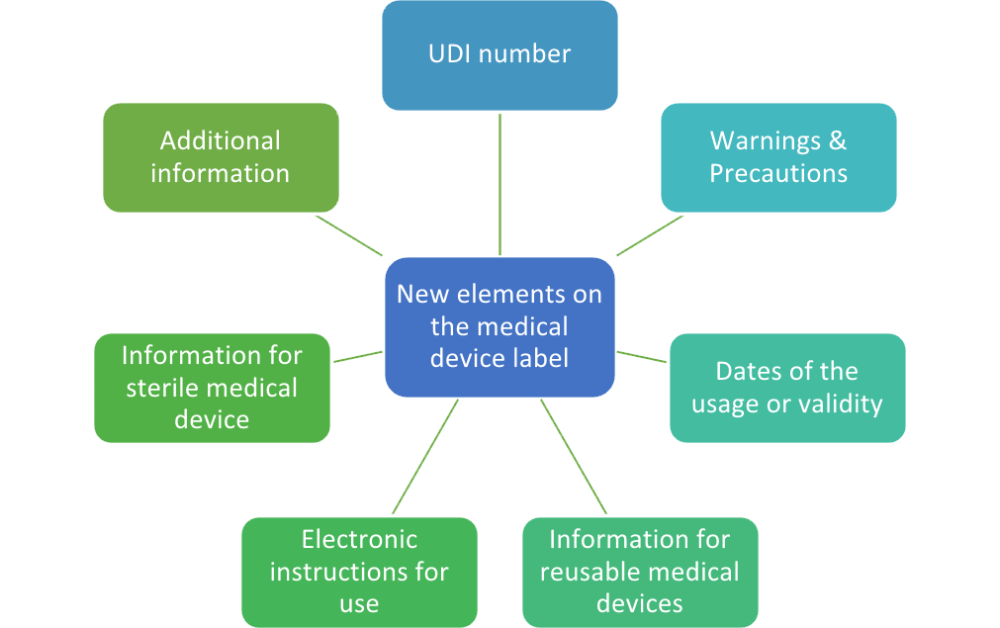
1) UDI 编号。
到目前为止,根据 MDD,必须在标签上包含批号和/或序列号。欧盟 MDR 引入了术语“UDI 编号”,它需要在标签上留出更多空间。除此之外,每个有源植入式设备都必须有自己唯一的序列号,而其他植入式设备则需要一个序列号或批号。
2)警告和注意事项。
附件 I 第 III 章第 23.2 节要求所有与器械有关的警告都必须印在标签上。但是,也声明可以将这些信息保持在最低限度,在这种情况下,使用说明中应出现更详细的信息,并考虑到预期用户。需要包含哪些警告的选择由制造商决定,但最好的方法是使用那些需要立即引起注意的警告。
3) 使用日期或有效期。
如果没有指明有效期,直到该医疗器械可以安全使用,制造日期必须出现在标签上。但是,该制造日期可以作为批号或序列号的一部分包含在内,前提是该日期可以清楚地识别。
4)可重复使用的医疗器械。
由于欧盟 MDR 为可重复使用的医疗器械引入了一个新类别,因此需要说明批准的再处理周期数以及任何限制。
5)电子使用说明。
可以放置一个可以找到电子使用说明的网址。这对于那些相当小的设备来说尤其方便,因为它们的物理空间不足,无法在标签上放置所有警告和注意事项。
6)无菌医疗器械。
除了所有其他必需的信息(无菌方法、无菌符号、有效期)外,现在还需要对无菌屏障系统的描述。包含此类符号的原因是为了降低无菌展示的特定风险,遵守源自欧盟 MDR 2017/745 的新法律要求,并提供额外的用户利益。

欧盟授权代表
7)附加信息。
医疗器械法规要求将所有其他具体信息进一步解释产品本身,以放置在标签上。这包括以下信息:
- 吸收率
- 血液或组织衍生物
- 纳米技术或计算机软件等创新
- 如果有药物或组织/细胞
- 存在致癌、致突变或生殖毒性或内分泌干扰物质
三、ISO 15223 的新修订版
这些增加的标签要求也反映在现有的 ISO 符号标准中。该标准有一个新的修订版: ISO/DIS 15223-1:2020 医疗器械 — 与医疗器械标签、标签和要提供的信息一起使用的符号 — 第 1 部分:一般要求 — 目前正在批准过程中。有涵盖新 MDR 要求的全新符号。
- 如何准备
实现 MDR 合规性自然会给医疗器械制造商带来标签挑战。公司需要确保其当前的标签系统适合其目的。因此,准备 MDR 标签的最佳方法是通过附件 I 一般安全和性能要求,第三章,要求 23.2 和 23.3,并找出其中哪些要求适用于您的医疗器械。定义之后,您需要使用 ISO 15223-1:2020 中的符号创建标签设计。不过,请记住,这种设计必须灵活,因为法规经常发生变化,您需要能够非常快速地响应这些变化。
以上就是关于“MDR标签是什么意思?”的相关内容,如果您想了解更多信息或办理MDR,请咨询在线客服或致电:400-106-2206。

医疗产品CE认证








