 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司欧盟MDR教程-欧盟MDR答疑

虽然欧盟MDR已经实施一段时间,但很多企业对它仍是一知半解,尤其是MDR的一些基础概念,经常有客户过来咨询相关事宜。今天小编根据大家的需求详细整理了一份欧盟MDR教程/欧盟MDR答疑,希望对大家有帮助~

欧盟MDR是什么意思?
欧盟MDR是欧洲议会和欧盟理事会于2017年发布的欧盟医疗器械法规 2017/745。欧盟MDR法规旨在确保在欧盟成员国生产或供应给欧盟成员国的医疗器械达到高标准的安全和质量。
该监管框架旨在通过欧盟数据库 (Eudamed) 更好地识别医疗器械,并标准化数据和技术进步。欧盟MDR法规旨在成为医疗器械的监管框架,可以可持续地确保健康和安全,同时仍然鼓励创新。
什么是MDD和MDR?
欧盟医疗器械指令 (MDD) 在被 2017 年发布的新欧盟医疗器械法规 (MDR) 取代之前已经实施了近 25 年。 该指令构成了欧盟医疗器械框架,但它不包括体外诊断医疗器械。
然而,确定对这些指令进行根本性修订是必要的,以建立一个更强大、透明、可持续和可预测的监管框架。其目的是提高医疗器械的整体健康和安全水平,同时仍支持行业创新。
欧盟MDR何时发布?
2017年4月4日欧洲医疗器械委员会负责制定新法规,而欧盟MDR法规于2017年5月5日发布。公司有一个三年的过渡期来遵守新法规MDR 欧盟法规。
欧盟MDR何时生效?
虽然公司可以立即开始遵守新法规,但从欧盟MDD指令到欧盟MDR法规的过渡期持续四年,并于2021年5月26日结束。在此日期之后,任何新的医疗器械都需要获得新的欧盟MDR法规认证。现有的MDD认证医疗器械有一个额外的过渡期,直到2024年5月,以更改其技术文档以符合新的MDR欧盟法规。

欧盟CE认证
所有现有的MDD认证设备必须在哪个日期根据MDR进行认证?
所有MDD认证设备在新 MDR 下获得认证的最后日期是2024年5月25日。如果在此日期之前医疗器械符合欧盟MDR法规,那么它可以根据MDR进行认证,但不是如果其MDD证书仍然有效,则为强制性。如果MDD证书在2024年5月25日之前到期,则此类医疗器械需要根据MDR进行重新认证。
根据欧盟 MDR 进行最终认证的最后日期是2024年5月25日——在此日期之后,所有投放市场的设备都必须根据欧盟医疗器械法规进行认证。

什么是欧盟MDR合规性?
欧盟医疗器械法规建立了一个类似于美国食品药品监督管理局(FDA)系统的唯一设备识别(UDI)系统。如果一家公司打算向欧盟市场提供或分销医疗器械,则这些产品上的标签需要符合这一新的UDI系统。对于打算在欧洲销售产品的医疗器械公司,必须遵守新的MDR法规。
什么是MDR认证?
医疗器械的MDR认证可验证该器械是否符合欧盟医疗器械的所有监管要求;该认证由CE标志表示。
为了使医疗器械获得认证,您的公司必须实施质量管理体系 (QMS)。许多公司使用ISO 13485作为实施此QMS的一种方式,因为这是欧盟协调清单上唯一的 QMS标准,因此它是实施与MDR欧盟法规相关的QMS的最佳方式。
与MDD相比,欧盟MDR有何变化?
欧盟医疗器械法规 (EU MDR) 取代了之前的欧盟医疗器械指令 (EU MDD)。特别是,新的欧盟 MDR (2017/745) 修改了之前的指令 2001/83/EC 并废除了理事会指令 90/385/EEC 和 93/42/EEC。
以下是旧欧盟MDD和新欧盟MDR的六大区别:
| 已更改的项目 | 欧盟MDR | 欧盟MDD |
| 产品分类 | 基于物质设备的新规则。 含有可被人体吸收物质的装置被纳入新的分类体系,导致许多装置被重新分类为更高风险等级。 (MDR第1、2、22、23、52条) |
设备分类系统。 |
| 临床评估过程 | 更严格的临床调查和上市后临床跟进领域。临床证据需要更新、清晰、令人信服并公开。 植入式医疗器械和III类器械的临床评估应至少每年更新一次。 (MDR第54、55、56、61至82条) |
需要进行临床评估。 |
| 公告机构 | 公告机构监管方面的重大变化。 通知机构将承担更多责任,有些机构可能会失去其作为通知机构的地位。 (MDR第35至50条) |
公告机构包括:由欧盟成员国指定对高级医疗器械进行评估的组织。 |
| Eudamed 变化 | Eudamed数据库补充:上市后监测、安全性和临床表现、定期安全性更新、更多临床调查数据和设备注册。 (MDR第33条) |
Eudamed数据库成立于2011年,是一个基于web的平台,用于存储监管机构信息。 |
| 经济经营者 | 为进口商、供应商、分包商、装配商和欧盟代表分配关税的指南。 (MDR第10、11、13.14、30条) |
未涉及 |
| UDI系统 | 独特的设备标识旨在增加耐多药医疗设备的可追溯性,特别是报告严重事件和识别假冒医疗设备。 | 未涉及在MDD中 |
什么是 MDR 和 IVDR?
欧盟 MDR 法规未纳入体外诊断法规 (IVDR);该 IVDR 设备受单独的欧盟法规 2017/746 管辖。但是,MDR 法规第 1 条(第 7 点)澄清了 IVDR 法规与欧盟 MDR 法规之间的关系:如果一个器械同时具有 IVDR 组件和其他医疗器械组件,则任何器械的 IVDR 部件均受 2017 /746 法规,但设备的其余部分受 MDR 欧盟法规的约束。

欧盟授权代表
体外医疗器械是用于诊断、监测或人体体外样本兼容性测量的器械。新的欧盟 MDR 法规包括对这些类型产品的要求。
MDR技术文件内容
与欧盟 MDD 的情况一样,所有具有 CE 标志的医疗器械产品都需要创建技术文件,以证明该器械符合欧盟指令对 CE 标志产品的所有要求。新欧盟 MDR 的附件 II 包括对内容、结构或技术文件中需要包含的内容的修订要求。
根据 EU MDR 的附件 II,技术文档需要清晰、有条理、易于搜索和明确。要包含在技术文件中的元素是:
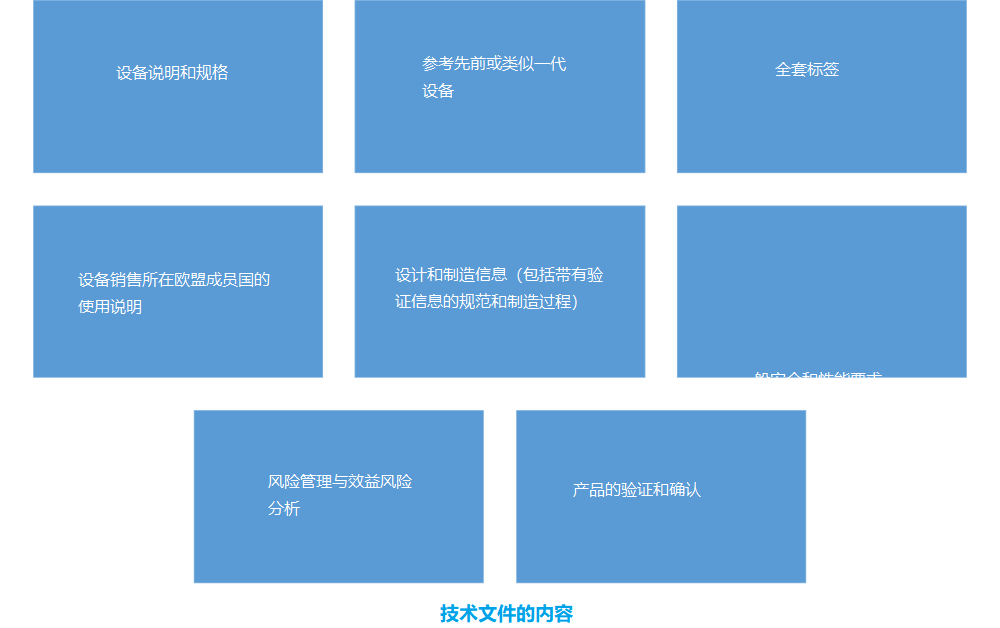
此做法与 ISO 13485:2016 条款 4.2.3 相匹配,该条款要求设备制造商创建技术文件或医疗设备文件。
什么是 I 类医疗器械?
I类医疗器械是被认为是非侵入性的医疗器械,包括以下类别:无菌、测量或可重复使用的手术设备。一些 I 类设备可以进行自我认证,不需要公告机构的参与;但是,您仍然必须实施 QMS。
医疗器械的例子有哪些?
欧盟 MDR 法规在附件八中包括五类医疗器械。本附件根据 22 条规则对产品进行分类。下面是一个简短的总结,每个都有一些例子:
- I 类(低风险):非无菌设备,或没有测量功能的设备。这些设备可以进行自我认证。例如:可重复使用的手术设备(如手术刀)、听诊器、轮椅。
- I 类特殊功能(低/中风险):无菌设备,或具有测量功能的设备。不符合其他类别要求的非侵入性器械,以及旨在暂时使用的侵入性器械。例如:再加工设备、测量设备、无菌交付的设备。
- IIa 类(中等风险):用于短期使用的侵入性设备。例如:牙齿植入物、气管切开插管、注射器、X 光设备。
- IIb 类(中/高风险):打算长期使用的侵入性设备。例如:植入式避孕、血袋。
- III 类(高风险):用于长期使用的植入式医疗器械。例如:起搏器、乳房植入物、椎间盘植入物、药物涂层支架。
什么是医疗保健中的 MDR?
欧盟 MDR 法规旨在提高欧洲医疗器械的安全性和性能,因此,旨在为这些医疗器械的患者和用户的健康提供高水平的保护。为此,欧盟 MDR 法规为医疗器械的质量和安全制定了高标准,以更好地解决医疗器械的安全问题。
那么,欧盟 MDR 与医疗保健的关系是什么?欧盟医疗器械法规正在制定一个框架,以确保制造或进口到欧盟的医疗器械符合这种高水平的安全和质量。通过提高医疗器械的安全性和性能,欧盟 MDR 法规旨在提高医疗保健的安全性。
以上就是关于“欧盟MDR教程-欧盟MDR答疑”的内容,如果您想了解更多信息或办理MDR,请咨询在线客服或致电:400-106-2206。

医疗产品CE认证








