 邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司
邮件群发-邮件群发软件|邮件批量发送工具|群发邮件平台|批量邮箱发送系统公司欧盟UDI与美国UDI的区别

UDI是医疗器械唯一设备标准符,它最终起源于美国,目的是产品的可追溯性,现在很多国家都在使用该编码。而欧美市场是中国企业非常热终的区域,今天小编就给大家讲解一下欧盟UDI与美国UDI的区别。
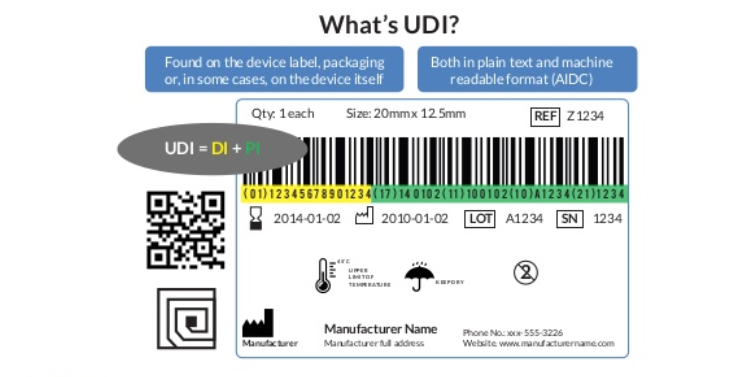
什么是UDI
唯一设备标识(UDl)旨在为美国境内的医疗设备分配一个唯一标识,它标记和标识各个医疗设备的整个分销和产品生活。生活UDl系统最初是创建的,由设备制造商根据全球设备识别标准进行开发和维护。现在,它也有助于采购和报销。
除某些例外情况外,UDI必须出现在医疗器械的标签上,并由两部分组成:
1. 设备标识符(DI)-UDI的一个强制性固定部分,用于标识设备的特定版本或型号设备;
2. 生产数据标识符(PI)-U Dl的条件可变部分,用于标识以下一项或多项。
当包含在设备,这将取决于制造商的内部质量体系。
- 生产日期
- 过期日期
- 生产批号
- 序列号
UDI=DI+PI
1. UDI历史
2007年,美国食品和药物管理局(FDA)开发了一种标签系统,可以唯一地识别市场上的每一种医疗器械(MD)。全球协调工作组(GHTF)很快认识到这种系统的全球相关性,并采纳了国际医疗器械监管论坛(IMDRF)于2013年发布的相应指南。由行业利益相关者和GHTF组成的监管机构的国际合作有意思的是,在美国市场经验丰富的医疗器械制造商很快认识到欧盟法规与美国食品和药物管理局(FDA)UDI指南的相似性。
随着医疗器械可追溯性处理的全球趋势,欧盟委员会在新的欧盟医疗器械法规(MDR)2017/745的最终文本中明确了实施独特器械识别(UDI)系统的要求。
欧盟UDI系统,如美国UDI要求,将分阶段实施。先从最高风险等级开始,最后从最低风险等级开始。

2. EUDAMED
EUDAMED将是一个信息系统,用于在欧盟委员会企业和行业总干事与欧盟成员国主管当局之间交换与欧盟医疗器械指令应用相关的法律信息。其法律依据在指令90/385/EEC、93/42/EEC、98/79/EC和2000/70/EC中均有规定。
根据这些指令,成员国需要确保投放市场并投入使用的医疗器械符合指令的所有规定,包括基本要求,不存在阻碍核武器自由行动的障碍设备指令还要求数据以标准化的格式存储到数据库中格式EUDAMED项目旨在解决指令中这一规定的有效实施问题。
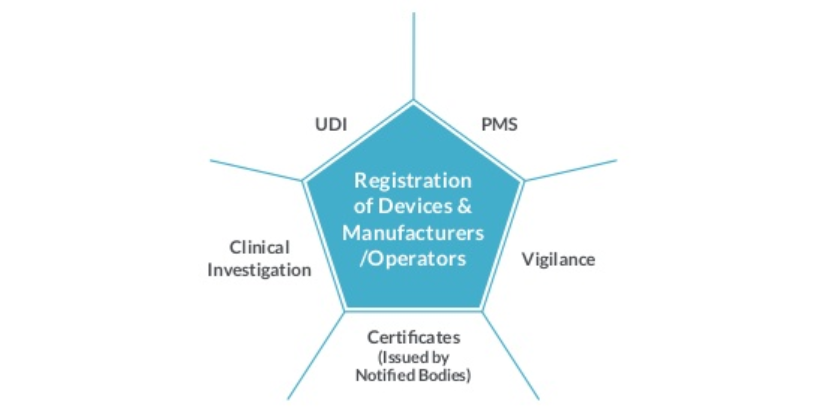
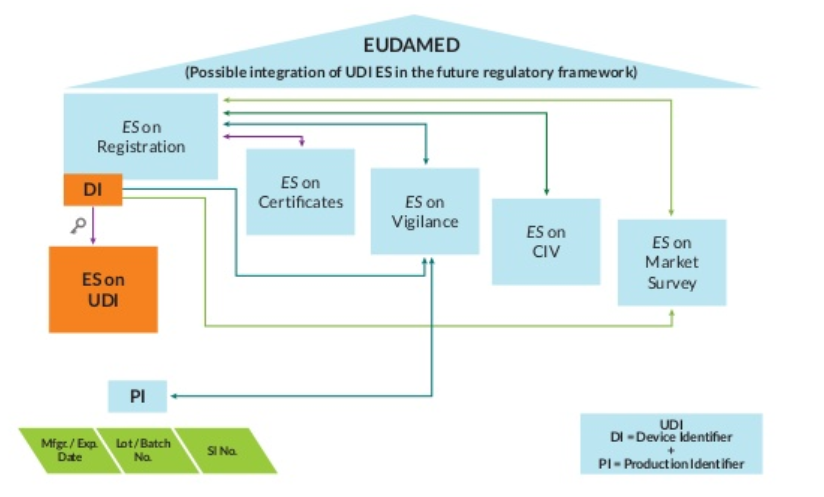
3. 欧盟EUDAMED美国GUDID的主要区别
EUDAMED将是一个信息系统,用于在欧盟委员会企业和行业总干事与欧盟成员国的主管当局之间交换与欧盟医疗器械指令应用相关的法律信息。指令90/385/EEC、93/42/EEC、98/79/EC和2000/70/EC规定了它的法律依据。
根据这些指令,成员国需要确保投放市场并投入使用的医疗器械符合指令的所有规定,包括基本要求,并且在批准的医疗器械自由移动方面不会遇到任何障碍设备指令还要求将数据存储到数据库中以标准化的方式格式EUDAMED项目旨在解决指令中这一规定的有效实施问题。
| 序号 | US GUDID | EU EUDAMED |
| 1 | 标签Duns编号 | 基本UDI-DI |
| 2 | 次要DI编号 | 单一注册号 |
| 3 | 直接标记(DM)装置,但豁免 | 如适用,授权代表的姓名和地址 |
| 4 | DM DI不同于主DI(和DM number) | 如适用,设备的附加商品名 |
| 5 | 客户联系人 | 设备的风险等级 |
| 6 | 处方使用(Rx)和/或非处方药(OTC) | 第26条规定的医疗器械命名代码 |
| 7 | 设备也是HCT/P套件和/或组合产品 | 如果适用,最大重复使用次数 |
| 8 | 上市前提交编号(PMA、补充编号510k或设备豁免) | 如适用,根据附录l第10.4.5节标记的信息 |
| 9 | FDA产品代码 | 其他信息的URL |
| 10 | FDA上市编号 | 如适用,严重警告或禁忌症 |
| 11 | GMDN编码 | 召回状态,启动现场安全纠正措施 |
以下是GUDID和EUDAMED之间的共同点,但可能需要翻译成24种欧盟官方语言:
- 名称或商品名
- 附加产品说明
- 临床规模
- 储存和搬运条件
- 设备的附加商品名
- 严重警告或禁忌症
欧盟UDI合规日期
与GUDID不同的是,EUDAMED对UDI采用了基于风险的方法提交。类III和植入式设备必须在2021年之前符合,lIa和IIb类设备必须在2023年之前符合,其中,对于IVD I类设备在2025年之前的实施也将基于风险,但IVDR的实施时间将有所不同。
D类设备应在2023年完工,C&B类设备应在2025年完工,而A类设备应在2027年完工。
*这些合规期限可能会发生变化,因为需求的实现取决于EUDAMED实施的进度和可用性。
以上就是关于“欧盟UDI与美国UDI的区别”的相关内容,了解更多点击咨询。

欧盟授权代表








